Disentangling homology, orthology, paralogy, identity and similarity with BLAST
BLAST, the first step to identify the evolutionary origin of proteins or genes
The Basic Local Alignment Search Tool (BLAST) is a powerful algorithm that helps researchers understand how different sequences are similar, including estimates of their evolutionary and functional relationships, and identification of gene family members. But, to really understand the relationships among gene or protein sequences, BLAST analysis is only the first step.
Following BLAST analysis, one way to define the relationship among protein gene sequences is by using phylogenetic analysis. This enables us to disentangle the evolutionary history of the genes, determining whether the studied sequences are orthologous, paralogous or homologous. Because there is some confusion around these terms, this post will try to explain the differences and give some examples.
Exploring the concepts of Orthology, Paralogy and Homology
Orthologous genes
Orthologs are genes derived from an ancestral gene that by speciation events diverge into different species (Jensen, 2001) (Figure 1). This is a simple definition, but it’s important to clarify two points:
- Orthologs originate from ancestral genes. If the gene in the ancestral genome has two copies as a result of gene duplication then orthology prediction is complicated. Gene duplication is a common mechanism for genes to evolve new functions. When a gene is duplicated, one copy can continue with its original function, while the other evolves a new function. Alternatively, the copy can become inactivated or non-functional. In either case the alterred copy becomes a paralog, retaining similarity with it’s ancestral gene.
- The definition specifies that the ancestral gene must be present in the last common ancestor of the compared species rather than in some arbitrary, more ancient ancestor.
*Orthologs are produced through the process of speciation by gene mutations and gene recombination. Frequently, orthologs retain the same biological function during the course of evolution.

Paralogous genes
Paralogs are genes that have arisen via gene duplication events. While recently duplicated genes can exist within the same genome, paralogous relationships also apply across species. and as such, they may or may not reside in the same genome (Figure 2) (Jensen, 2001). This definition involves some important points:
- The age of the duplication event leading to the emergence of paralogs can vary widely. Many of these duplications may have occurred early in life’s evolution.
- Recent paralogs may perform related but distinct functions (e.g., performing similar metabolisation of different substrates), but older paralogs often have biologically distinct functions. This potential functional divergence is much higher than for orthologs.
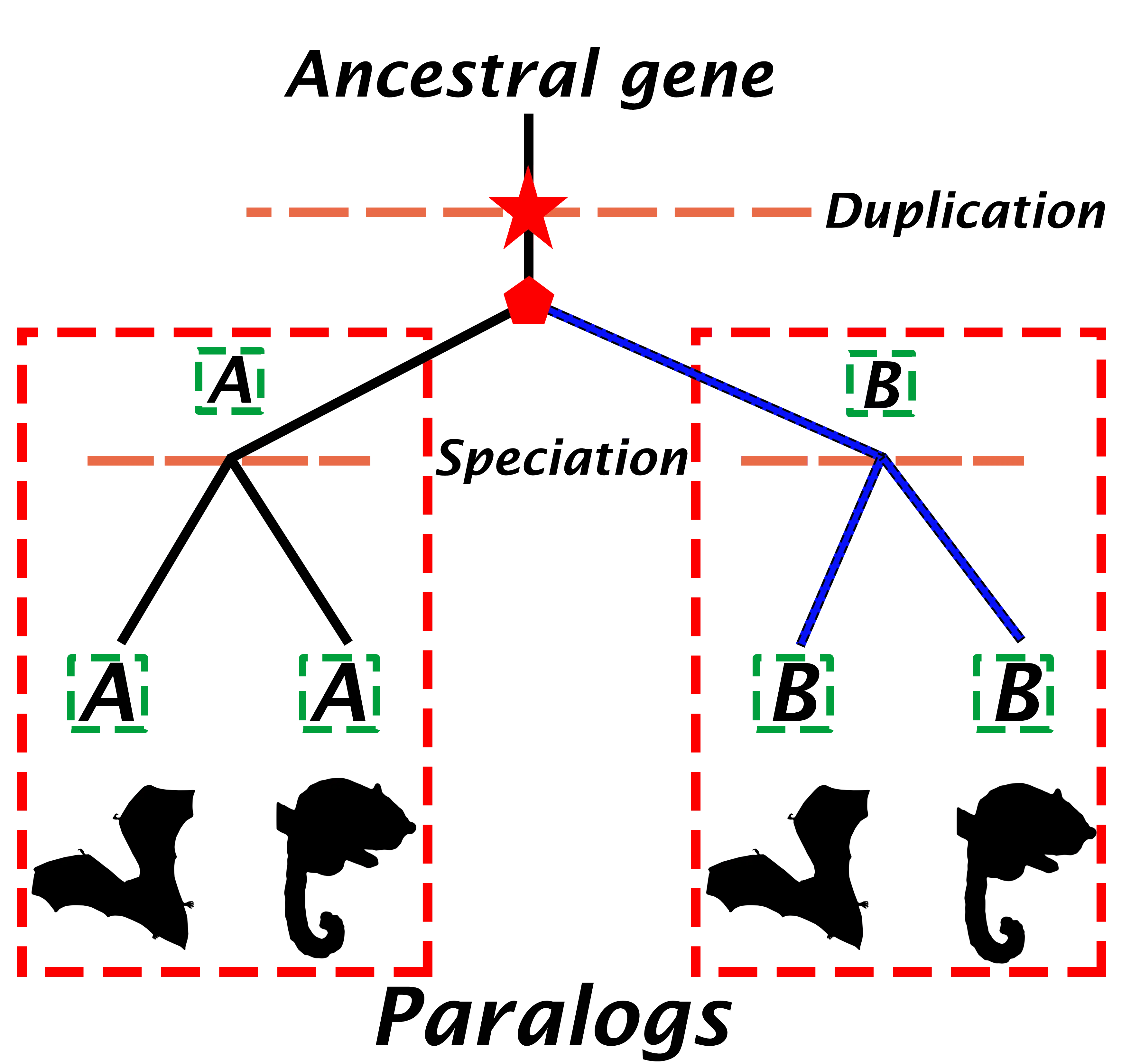
Homologous genes
Homology refers to morphological structures that have arisen through common descent. For example, the forelimbs of all mammals are homologous, derived from a fish-like common ancestor, but the forelimbs of mammals and insects are not homologous because they were independently derived. Homology applies to genes as well as individual nucleotides in the same way: they are genes with a evolutionary common origen, made up of nucleotidesthat also share a common origin. This origin can be traced back to an ancestral gene that existed in a common ancestor of the compared species. The main point is:
Orthologs and paralogs are different types of homologs. Homology is a binary definition: either two sequences are homologous or they are not homologous. There is no percentage of homology.
Hemoglobin as an example of Orthology, Paralogy and Homology
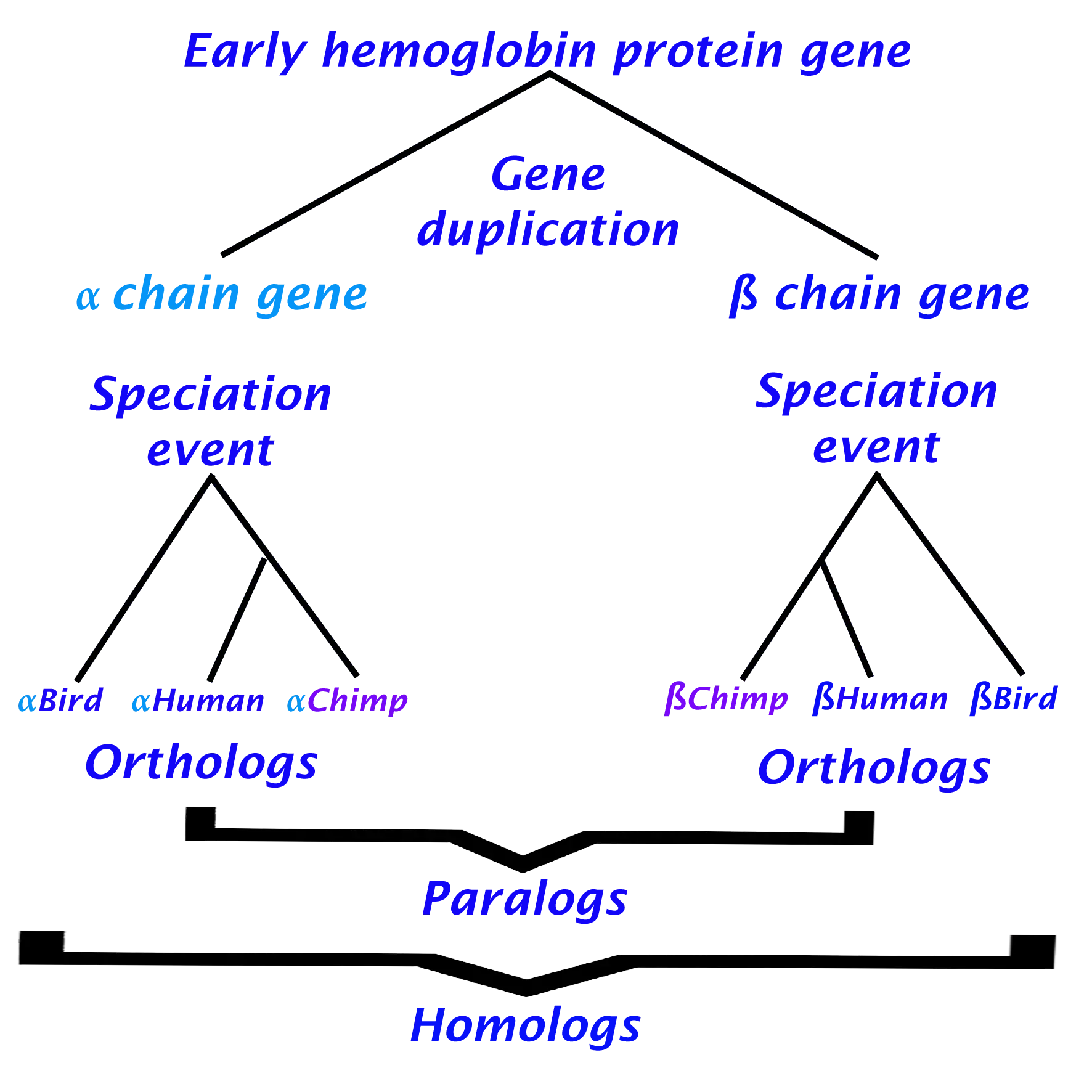
Hemoglobin is a protein found in red blood cells and is responsible for transporting oxygen from the lungs to the tissues and removing carbon dioxide from the tissues. In humans, hemoglobin is composed of two alpha chains and two beta chains, which have slightly different amino acid sequences and distinct properties. Specifically, the beta chains are more sensitive to changes in oxygen concentration. Alpha and beta hemoglobin chains are believed to have originated from a gene duplication event and subsequently diverged through speciation (Wideman et al., 2018).
Analyzing the previous information we can extract the following:
- As a result, of gene duplication the hemoglobin ancestor gene diverged into two genes alpha and beta.
- After duplication, a speciation event creates several copies of the alpha gene distributed in different species as well as for the beta gene
- Alpha and beta genes have different functions
Therefore we can conclude that hemoglobin alpha and beta genes code for paralogous proteins, whereas alpha hemoglobins across different mammalian species are orthologous, just as beta homoglobins are orthologous (but alpha and beta are paralogous to one another Figure 3).
Identity and similarity
Identity and similarity are important concepts that are commonly used in sequence alignment methods like BLAST.
Identity
Identity refers to the percentage of exact nucleotide or amino acid matches between two sequences (Figure 5), where the nucleotide or amino acid in one sequence matches the nucleotide or amino acid in the other sequence. For example, if two DNA sequences of the same length have 90% identity, it means that 90% of their nucleotides are the same (Korf et al., 2003).
Similarity
Similarity refers to the percentage of matches between two sequences, taking into account both exact matches and conservative substitutions Figure 4 and Figure 5. Conservative substitutions are amino acid substitutions that are less likely to affect protein function. For example, replacing a hydrophobic amino acid with another hydrophobic amino acid is considered a conservative substitution. Substitutions that don’t change a gene’s function are much more common than those that do, so downweighting changes that don’t alter function allows programs to align more distantly related genes. Similarity is usually calculated using a scoring matrix, such as the BLOSUM matrix, which assigns scores to different types of amino acid substitutions based on their likelihood of occurring in nature (Korf et al., 2003).
In summary, identity and similarity are important measures of sequence conservation and are used in sequence alignment tools like BLAST to compare DNA or protein sequences. While identity measures exact matches, similarity also takes into account substitutions based on probability.
Warning: Be careful interpreting BLAST’s identity scores. This is because BLAST reports % identity for specific aligning parts, typically not for the entire query or hit sequence. Imagine a pair of homologous sequences, 300 nucleotides long, that are highly diverged for the first 100 and the last 100 nucleotides. If you query a sequence from the middle 100 nucleotides, BLAST may report 100% identity, but this cannot be extended to the entire 300 nucleotides.
Is sequence similarity and identity a good approach to predicting the evolutionary origin of proteins and genes?
Using similarity alone to determine homology can be problematic because it does not take into account the possibility of convergent evolution, functional convergence, mutation rate or gene conversion (Pearson, 2013).
- Convergent evolution refers to the independent evolution of similar traits in distantly related organisms. It implies that some solutions or structures are better than others, which puts selective pressure on any organism to “converge” on this solution.
- Functional convergence refers to the evolution of similar functions in unrelated genes due to similar selective pressures. Mutation rate is the number of new mutations in each generated by the probability of each new mutation reaching fixation. Gene conversion refers to the unidirectional transfer of genetic material from a “donor” to an “acceptor” sequence. It mediates the transfer of genetic information from intact homologous sequences to regions containing double-strand breaks (DSBs), and can occur between sister chromatids, homologous chromosomes, and even between homologous sequences on the same chromatid or on different chromosomes. Gene conversion occurs predominantly in meiosis, but also in mitosis.
In practice, this means that sequences that are similar today may have been less equal in the past.
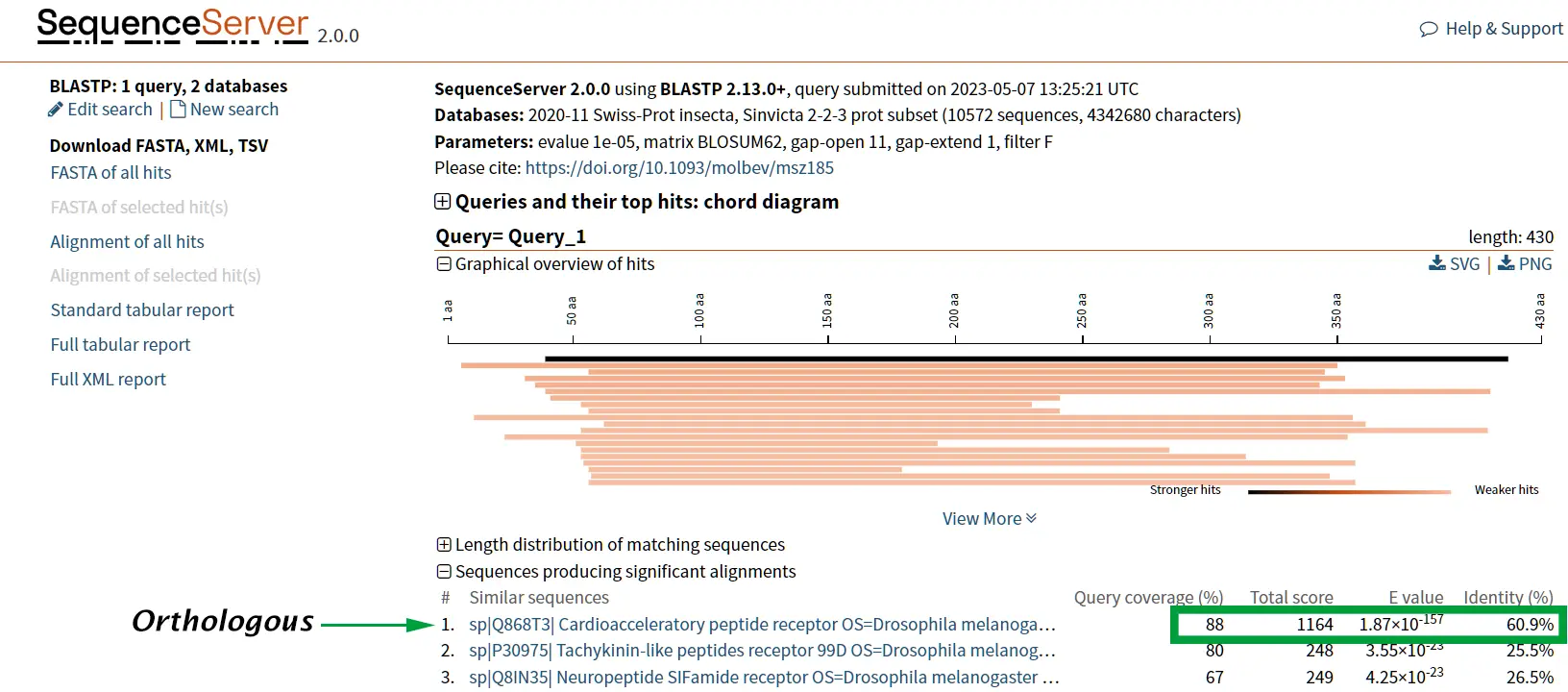
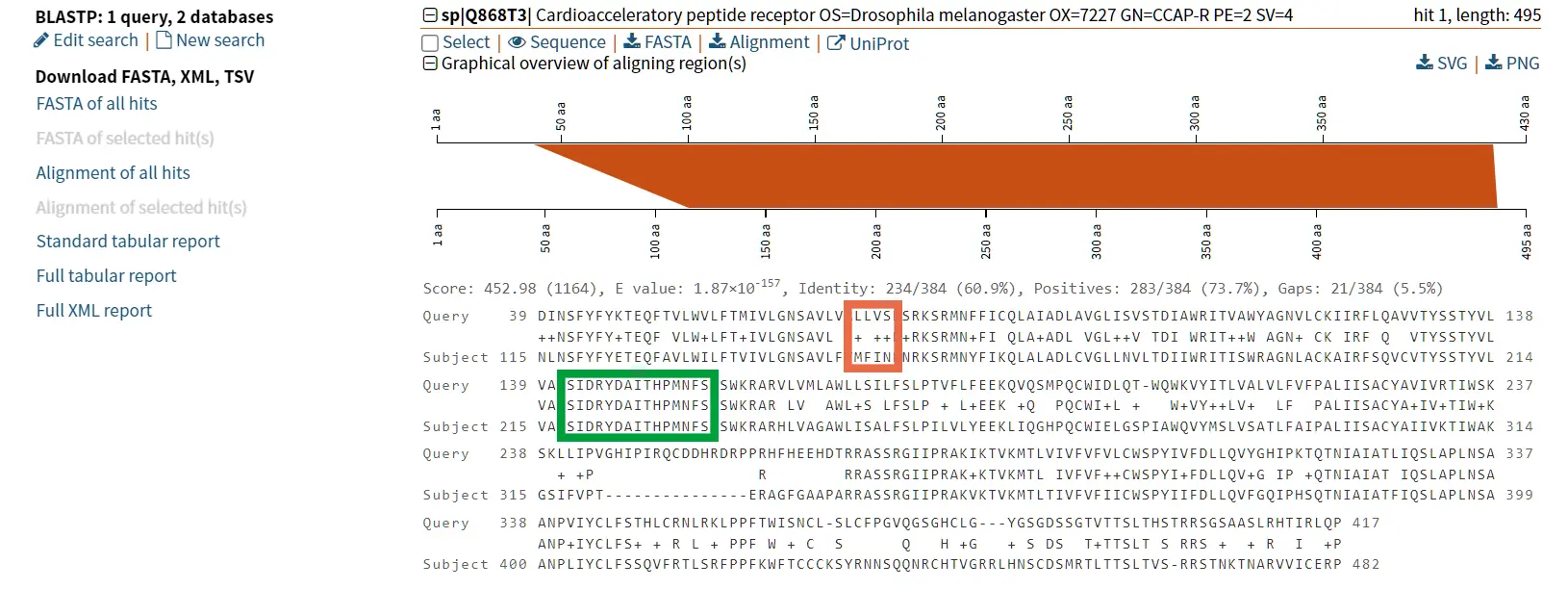
Case study: Tachykinin and neuropeptide Y
The proteins tachykinin and neuropeptide Y (NPY) were called structurally similar due to their high degree of sequence similarity (Figure 6 and 7) (Larhammar et al., 1992). If we compare the scores of Tachykinin-NPY alignment with the scores of the first hit of the BLAST results (Neuropeptide F) we can observe that the values are simmilar (Figure 6). Due to the Neuropeptide F, which is considered the prostomian ortholog of neuropeptide Y, we can think that tachykinin and neuropeptide Y are also closely related.
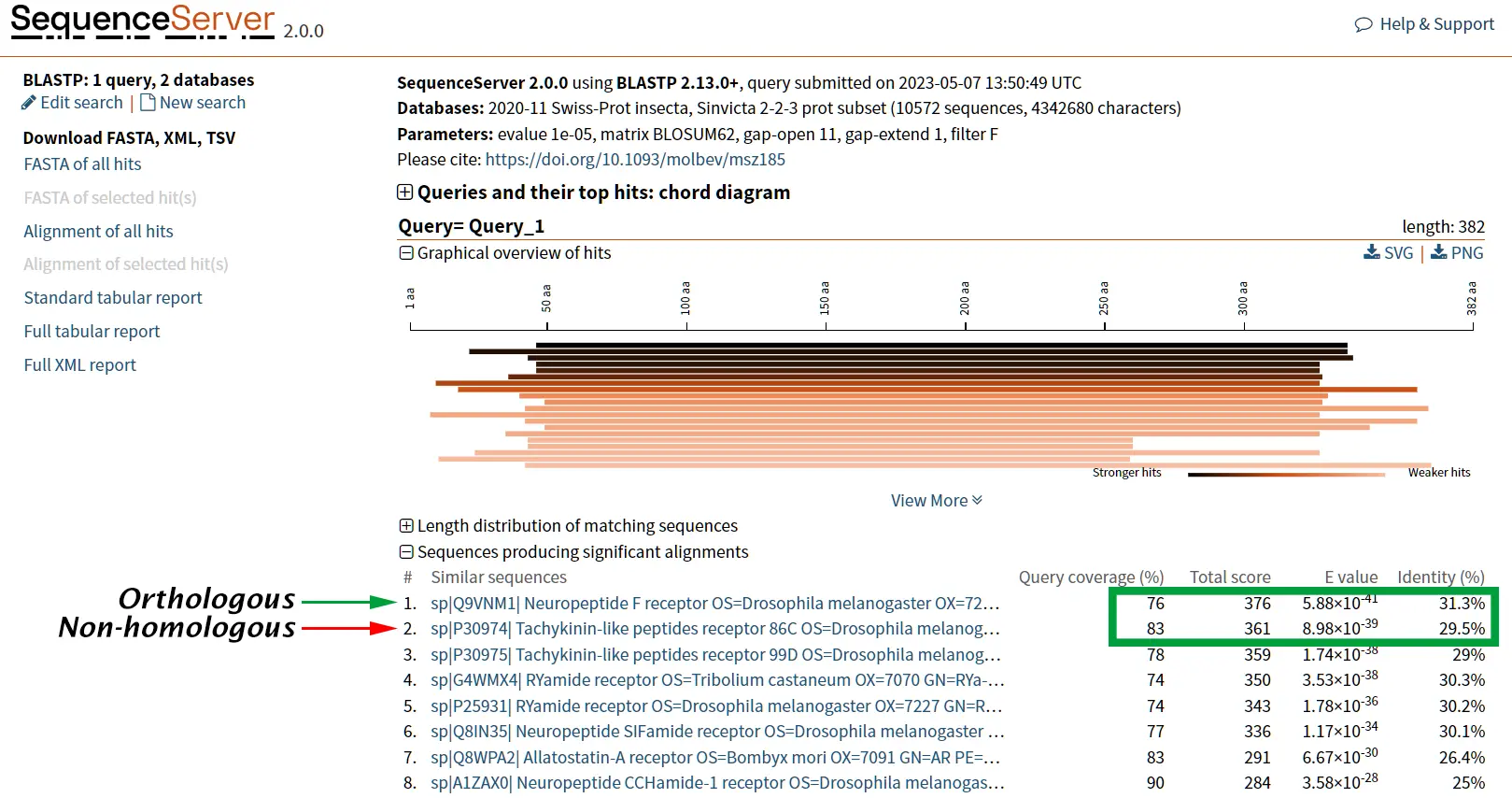
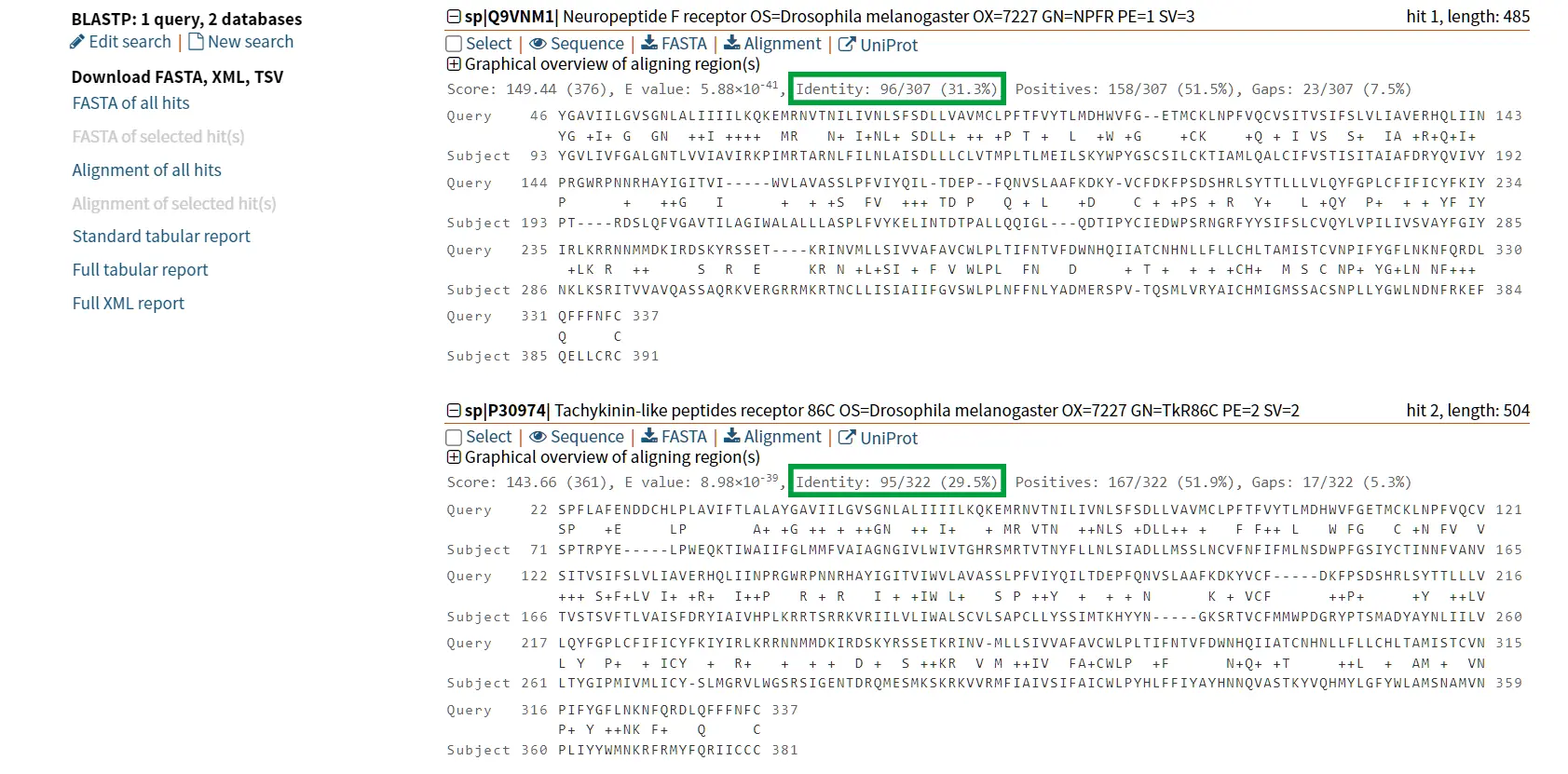
However, it is known that Tachykinin peptides and NPY are not closely related proteins and that their similarities could be the result of convergent evolution. Thus, while sequence similarity is a useful tool for identifying potentially homologous sequences, additional lines of evidence are needed to confirm homology and rule out other explanations for observed similarities.
Then to elucidate the evolutionary relationship among proteins or gene sequences, biologists use a variety of approaches to infer homology:
- Phylogenetic analysis, which estimates the evolutionary relationships of different genes or proteins across multiple species.
- Analysis of conserved motifs or domains across multiple species.
- Examination of gene structure.
- Synteny, which compares the order and arrangement of genes or genomic regions across different species.
Thus, by combining multiple lines of evidence, biologists can more confidently infer homology and better understand the evolutionary relationships among different genes and proteins (Koonin, 2005).
What can we conclude?
- Orthology, paralogy, homology, sequence similarity and identity are based on similar principles of shared ancestry and evolutionary relationships among genes and proteins, they differ in the details of their definitions and applications.
- Orthology refers to genes or proteins that share a common ancestry (homology) and have diverged through speciation events, while paralogy refers to genes or proteins that have arisen through gene duplication events. Homology, on the other hand, is a broader concept that encompasses both orthology and paralogy. Both orthologs are paralogs are homologous, in that they were derived from common ancestry, but the common ancestor of an ortholog is more recent than the common ancestor of a paralog.
- Sequence similarity and sequence identity are commonly used as proxies for assessing homology, but they do not always indicate homology and must be interpreted with caution.
- Functional and structural conservation, gene synteny, and phylogenetics, can help to confirm or reject homology.
Therefore, a comprehensive understanding of the concepts of orthology, paralogy, and homology, as well as their relationship to sequence similarity and sequence identity, is essential for the accurate interpretation of evolutionary relationships between genes and proteins.
References
Jensen, R. A. (2001). Orthologs and paralogs-we need to get it right. Genome Biology, 2(8), interactions1002–1.
Koonin, E. V. (2005). Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet., 39, 309–338.
Korf, I., Yandell, M., & Bedell, J. (2003). Blast. “ O’Reilly Media, Inc.”.
Larhammar, D., Blomqvist, A., Yee, F., Jazin, E., Yoo, H., & Wahlested, C. (1992). Cloning and functional expression of a human neuropeptide y/peptide YY receptor of the Y1 type. Journal of Biological Chemistry, 267(16), 10935–10938.
Pearson, W. R. (2013). An introduction to sequence similarity (“homology”) searching. Current Protocols in Bioinformatics, 42(1), 3–1.
Wideman, J. G., Balacco, D. L., Fieblinger, T., & Richards, T. A. (2018). PDZD8 is not the ‘functional ortholog’of Mmm1, it is a paralog. F1000Research, 7.